


Investigadores del CIBER-BBN del grupo de ‘Biología y Fisiología Celular-LABRET’ de la Universidad de Málaga han descrito una nueva enfermedad genética del esqueleto mediante una estrategia de medicina de precisión.
Utilizando métodos de secuenciación masiva -de todos los genes- han identificado las mutaciones que causaban un tipo de patología ósea rara, en concreto, las del gen ‘LAMA5’, encargado de codificar una proteína de matriz celular que rodea vasos sanguíneos en tejidos esqueléticos.
Esta enfermedad consiste en una fragilidad ósea extrema con falta de mineralización y deformación esquelética asociada con dislocación en articulaciones y cardiopatías, así como una deficiencia pulmonar que provoca letalidad perinatal -en el momento del nacimiento-.
El estudio se ha realizado desde el Centro Andaluz de Nanomedicina y Biotecnología (BIONAND), en colaboración con el Registro Internacional de Displasias Esqueléticas de la Universidad de California (Los Ángeles), donde se desarrolló la secuenciación de los genes de pacientes con esta enfermedad genética. También ha participado la Universidad Masaryk, de la Republica Checa.
“Nuestro equipo científico lleva años investigando síndromes genéticos raros que afectan al esqueleto con el fin de dar solución médica a pacientes con difícil diagnóstico y tratamiento”, explica el investigador del Departamento de Biología Celular, Iván Durán, autor principal de este estudio, cuyos resultados han sido publicados en la revista científica ‘EBIOMEDICiNE’.
Según el experto, la medicina de precisión es la llave para descubrir qué factores genéticos y moleculares provocan este tipo de patologías y, por tanto, entender el mecanismo que las causa y poder desarrollar terapias personalizadas.
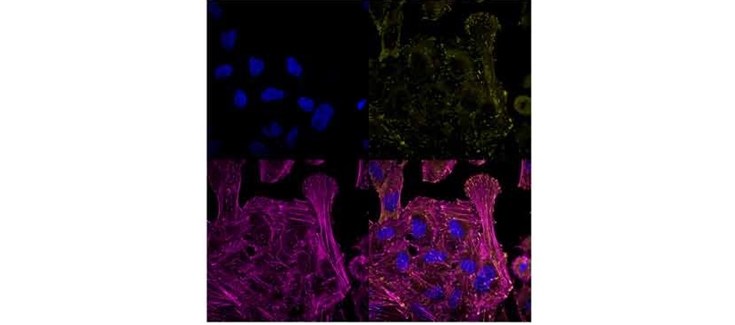
Así, los investigadores de la Universidad de Málaga han descrito también el mecanismo de enfermedad generando modelos celulares por edición genética imitando las mutaciones en ‘LAMA5’, con el objetivo de confirmar si estas son el origen y conocer el proceso molecular que desencadena el problema. Estos modelos celulares se han generado por edición genética con CRISPR, introduciendo mutaciones que causasen un gen nulo o hipomorfo.
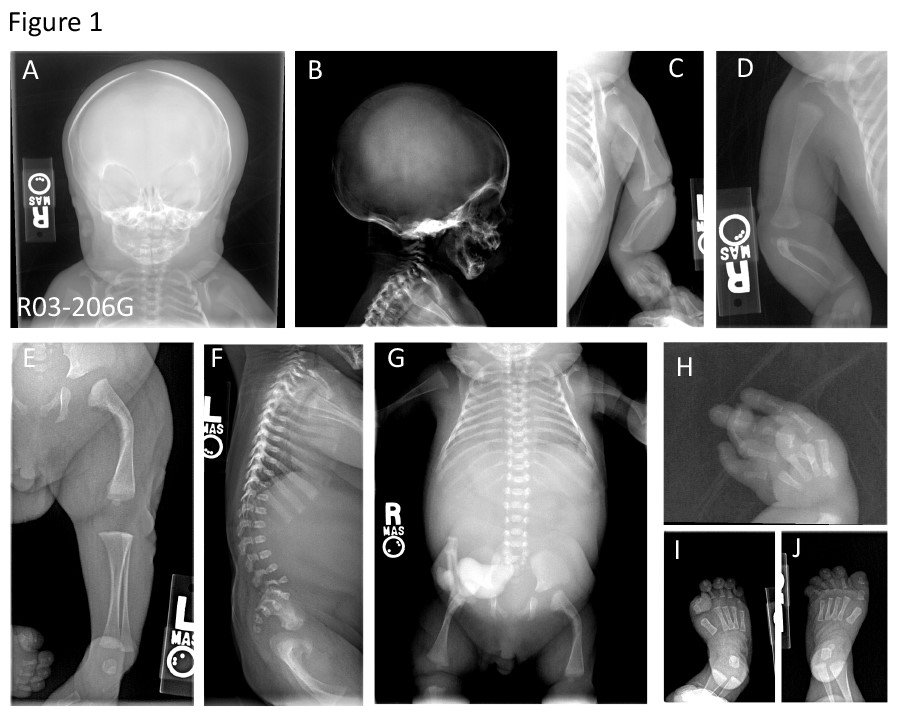
Estudio radiográfico de pacientes con mutaciones el LAMA5 con defectos de mineralización generalizada en cráneo y huesos largos, curvatura medial en diáfisis, deformaciones en costillas, vertebras y luxaciones y contracciones en articulaciones
“Gracias a estos modelos averiguamos una nueva ruta de señalización que gobierna la formación del esqueleto –para que el hueso crezca y se mantenga sano-, lo que significa que nuestro trabajo no solo ha llevado al descubrimiento de una nueva enfermedad, sino a un mecanismo inédito que puede ser explotado para afecciones óseas comunes”, afirma Durán.
Tal y como aclara el investigador, la presencia de ‘LAMA5’ entre células que dirigen la formación de esqueleto indica, por tanto, que la aparición de señales provenientes de vasos sanguíneos especiales puede ser un arma muy efectiva para la reparación y regeneración ósea.
“Los vasos sanguíneos no solo proveen de irrigación al hueso, sino que también transportan señales y albergan nichos de células madre que pueden ser movilizadas para inducir un proceso regenerativo. ‘LAMA5’ parece ser un componente clave para albergar células madre del tipo pericitos”, aclara.
La osteoporosis y la osteogénesis imperfecta son enfermedades que causan debilidad ósea y que afectan a un porcentaje muy significativo de la población. Además, estas patologías suelen presentar defectos óseos que se reparan con gran dificultad. Este nuevo hallazgo científico permitirá diseñar nuevos tratamientos y estrategias para todo tipo de afecciones de fragilidad ósea.
En este sentido, junto con la red CIBER-BBN y la Red de Terapia Celular, el grupo ‘Biología y Fisiología Celular’ de la UMA, que también forma parte de IBIMA, avanza en un nuevo trabajo para desarrollar un biomaterial osteogénico que sirva para curar fracturas complejas en personas con fragilidad ósea y baja capacidad regenerativa.
*Imagen: Estudio radiográfico de pacientes con mutaciones el LAMA5 con defectos de mineralización generalizada en cráneo y huesos largos, curvatura medial en diáfisis, deformaciones en costillas, vertebras y luxaciones y contracciones en articulaciones
Artículo de referencia:
Barad M, Csukasi F, Kunova-Bosakova M, Martin J, Zhang W, Taylor SP, Dix P, Lachman R, Zieba J, Bamshad M, Nickerson D, Chong JX, Cohn DH, Krejci P, Krakow D, Duran I. Mutations in LAMA5 disrupts a skeletal noncanonical focal adhesion pathway and produces a distinct bent bone dysplasia. 2020 EBioMedicine. Nov 23;62:103075. doi: 10.1016/j.ebiom.2020.103075




Av. Monforte de Lemos, 3-5. Pabellón 11. Planta 0 28029 Madrid




